
VirMiner is a software tool which provides comparatively comprehensive phage information in metagenomic data: 1) identifies phage contigs using reliable pre-trained model; 2) gets full functional annotation for these phage contigs; 3) predicts possible phage-host relationships using existing tools; 4) if users upload two groups of metagenomic samples, downstream analysis for comparison among different groups would be done. Visit about or help page to see more details.
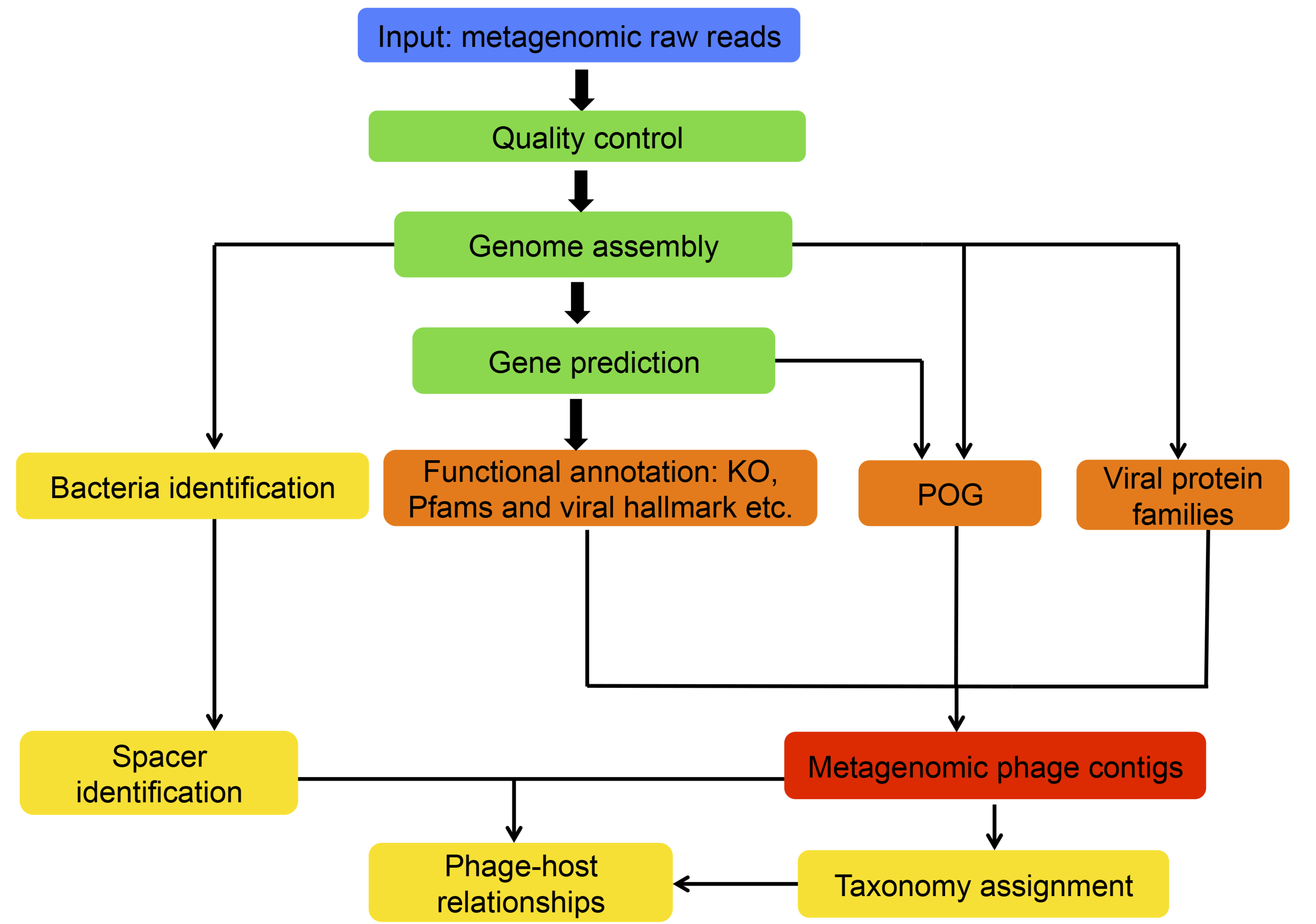
The VirMiner analysis pipeline
The data processing steps include: Sequence raw reads uploaded by users are quality controlled by removing adapter sequence, duplication reads and low quality reads. The high quality reads are assembled into contigs. Genes are predicted based on these assembled into contigs.
Functional profiles are generated by mapping protein sequence to different databases including KO, Pfam and viral hallmark genes defined by Roux et al., viral protein families identified in Paez-Espino et al., Phage Orthologous groups (POG) both orgininally developed by Kristensen et al. and recently updated by ourselves. These functional profiles are severed as the metrics involved in random forest model to identify metagenomic phage contigs.
Predicting phage-host relationships using CRISPR-spacer based method. CRISPR recognition tool (CRT) is employed to identify spacers from bacteria then all identified spacers were queried for sequence matches against metagenomic phage contigs by blastn.
Inter-group comparison (optional)
If users upload metagenomic samples under two conditions ("Control" and "Treatment"), Differential abundance analysis (at the level of Pfam or KO) can be performed.